
When the FDA shows up at your generic drug manufacturing facility, it’s not a surprise visit-it’s a CGMP compliance check. And if you’re not ready, it can turn into a costly, time-consuming ordeal. The FDA doesn’t inspect randomly. They use a risk-based system that looks at your history, your products, your complaints, even tips from insiders. If you make generic medicines for the U.S. market, you’re on their radar. Here’s exactly what happens during an inspection-and how to get through it without a warning letter.
What the FDA Is Really Looking For
The FDA doesn’t just check if your machines are clean. They’re evaluating your entire quality culture. Are your people trained? Do your procedures match what you told them in your application? Are your records real-or were they backfilled after the fact?
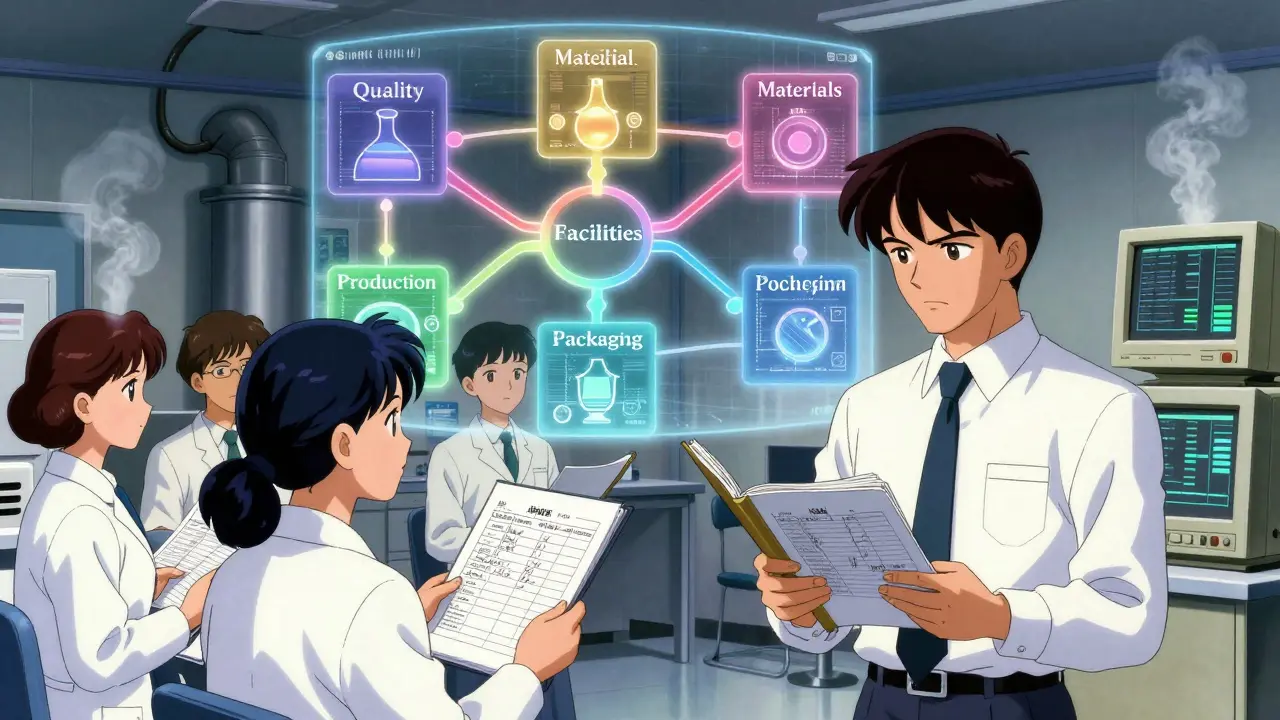
They use a 6-system approach:
- Quality System-always inspected. This includes your quality unit, deviation handling, CAPAs, and change control.
- Facilities & Equipment-are your cleanrooms maintained? Is your equipment qualified and calibrated?
- Materials-do you test every incoming raw material? Are your suppliers approved?
- Production-does your process match your validation? Are batch records accurate?
- Packaging & Labeling-is the right label on the right bottle? Are lot numbers traceable?
- Laboratory Control-are your methods validated? Are your stability samples stored correctly?
The Quality System is the backbone. If it’s weak, everything else falls apart. The FDA knows this. They start here-and they’ll dig deep. You’ll be asked for your quality unit’s charter, training records, and examples of how they’ve stopped a bad batch before it shipped.
The Inspection Process: From Door to Report
Inspections can be announced-or not. The FDA can show up unannounced, especially if you’ve had past issues or if there’s a spike in complaints. Don’t assume you’ll get a heads-up.
When they arrive, they’ll ask for your inspection room. This isn’t just a conference room. It needs to be quiet, clean, and stocked with everything they might ask for: SOPs, batch records, training logs, validation reports, equipment logs, and stability data. Bring copies. Bring originals. Bring backups.
The inspectors will walk your facility. They’ll watch your operators. They’ll ask questions like:
- “Show me how you handle a deviation.”
- “Why did this batch fail the dissolution test?”
- “Who approved this change to the mixing time?”
- “Where are your stability samples from batch #G2025-048?”
They’ll pull records. Not just one or two. They’ll pick random batches and trace them from raw material to finished product. If your records don’t tell a clear, consistent story, they’ll note it.
At the end, they’ll hand you Form FDA 483. This isn’t a final verdict. It’s a list of observations-things they saw that don’t meet CGMP standards. They list them in order of seriousness. A missing signature on a training log? Minor. A batch released without full testing? Major.
What Happens After the FDA Leaves
You have 15 business days to respond to the FDA 483. This is your chance to fix things before it becomes a warning letter.
Your response needs to be specific. Not “We’ll train our staff.” But “We’ve updated SOP 7.3 on deviation reporting, trained all 14 production staff on January 10, 2026, and attached the training attendance sheet and quiz results.”
The FDA doesn’t just read your response. They cross-check it. They’ll look at your CAPA system. They’ll see if you’ve fixed the same issue before. They’ll check if your quality unit was involved. If your response is vague, delayed, or doesn’t match your records, they’ll issue a warning letter.
Warning letters mean public scrutiny. They mean delayed approvals. They mean more inspections. And if you don’t fix it? The FDA can refuse to approve your new products-or even block your existing ones from entering the U.S. market.
Pre-Approval Inspections (PAIs): The Make-or-Break Moment
If you’re submitting a new generic drug application, expect a Pre-Approval Inspection. This is your final checkpoint before the FDA says yes.
They’ll ask three questions:
- Is your facility ready for commercial production?
- Does what you wrote in your application match what you’re actually doing on the floor?
- Is your data complete and accurate?
They’ll compare your application’s process description to your validation reports. They’ll check if your analytical methods match what you filed. They’ll pull stability samples and verify storage conditions. If your application says “stored at 25°C/60% RH,” but your stability chamber is at 28°C? That’s a red flag.
PAIs are the most stressful inspections. One major finding can delay approval by 12-18 months. That’s millions in lost revenue.
The New PreCheck Program: A Game Changer
In 2024, the FDA launched PreCheck-a program designed to help manufacturers avoid problems before they happen. It’s not a shortcut. It’s a roadmap.
Through PreCheck, you can submit a Type V Drug Master File (DMF) early in your facility’s design phase. You share your layout, your quality system, your equipment plans. The FDA reviews it and gives feedback. You fix issues before you spend millions building a facility that won’t pass inspection.
Companies using PreCheck report smoother PAIs and fewer FDA 483s. Why? Because they’re not guessing what the FDA wants. They’re building to the standard from day one.

What Successful Facilities Do Differently
The FDA says over 90% of inspections find acceptable compliance. So what separates the 90% from the 10%?
It’s not fancy equipment. It’s not big budgets. It’s three things:
- Constant readiness-they don’t prep only when they hear the FDA is coming. They operate like they’re always being inspected.
- Clear documentation-every action has a record. Every decision has a traceable reason.
- Quality ownership-the quality unit isn’t a paperwork department. They have authority to stop production. And they use it.
One facility in Ohio had zero FDA 483s for five years. Why? They ran monthly mock inspections. They trained every new hire on what an FDA inspector might ask. They kept their SOPs simple and updated them quarterly. They didn’t wait for problems-they prevented them.
How to Prepare: A Practical Checklist
You don’t need to be perfect. But you need to be consistent. Here’s what to do now:
- Review all your SOPs. Are they written in plain language? Are they followed?
- Check your deviation logs. Are you fixing the same issues over and over?
- Verify your equipment qualification records. Are they current? Are they signed?
- Test your analytical methods. Are they validated? Are they documented?
- Locate your last 10 stability samples. Can you show where they’re stored and when they were tested?
- Train your staff. Not just on procedures-on how to talk to inspectors.
- Run a mock inspection. Bring in someone from another site to play FDA.
- Designate your inspection room. Stock it with everything they might need-today.
Don’t wait for the letter. Start now. The FDA doesn’t reward last-minute fixes. They reward consistent quality.
What the FDA Won’t Tell You
The FDA isn’t trying to shut you down. They want you to make safe, effective medicines. But they’re under pressure-to protect patients, to keep up with global supply chains, to prevent shortages.
They’re watching for data integrity. If your records look too clean, if signatures are missing, if timestamps don’t add up-they’ll dig. They’ve seen it all.
And they’re watching for culture. Do your employees speak up when something’s wrong? Or do they hide mistakes? The FDA can tell the difference.
Be honest. Be thorough. Be ready.
Can the FDA inspect my facility without warning?
Yes. The FDA can conduct unannounced inspections, especially if your facility has a history of compliance issues, receives consumer complaints, or is flagged by their risk-based system. You should always operate as if you’re under inspection.
What’s the difference between an FDA 483 and a warning letter?
An FDA 483 lists observations from an inspection-it’s not a final decision. A warning letter is a formal regulatory action issued if the FDA believes your responses to the 483 are inadequate or if violations are severe. Warning letters are public and can block product approvals.
How long does a typical FDA inspection last?
Most inspections last 3 to 7 days, depending on facility size and complexity. A small facility with a single product line might be done in 2-3 days. A large, multi-product site with complex processes can take a week or more.
What’s the most common reason for an FDA 483 in generic manufacturing?
The most frequent observation is failure to properly investigate deviations and implement effective CAPAs. Other top issues include inadequate validation of manufacturing processes, poor data integrity, and lack of documented training for key personnel.
Does the FDA inspect facilities outside the U.S.?
Yes. The FDA inspects foreign facilities that manufacture drugs for the U.S. market. In fact, more than half of generic drug manufacturing facilities inspected by the FDA are located outside the United States. The same CGMP standards apply regardless of location.
What is the PreCheck program, and should I use it?
The PreCheck program lets manufacturers submit facility and process details to the FDA early-during design or construction. The FDA reviews it and gives feedback before you invest in full production. If you’re building a new facility or launching a new product, PreCheck can save you millions by avoiding costly redesigns after an inspection failure.
Shanna Sung
January 3, 2026 AT 19:14The FDA is just a puppet for Big Pharma. They’re not inspecting for safety-they’re protecting patent monopolies. Every time they hit a generic maker with a 483, it’s because someone’s stock price dipped. I’ve seen the emails. They’re coached to find flaws. Always.
Allen Ye
January 5, 2026 AT 03:13What’s fascinating here isn’t the checklist-it’s the underlying philosophy. The FDA’s 6-system model isn’t regulatory bureaucracy; it’s a mirror of organizational epistemology. Are your records real? That’s not a compliance question-it’s a question of whether your institution believes truth matters. If your quality unit can’t stop a batch, you’ve outsourced ethics to a spreadsheet. The real failure isn’t a missing signature-it’s the cultural surrender to efficiency over integrity. And that’s why foreign plants fail: not because of equipment, but because leadership treats compliance as a cost center, not a moral imperative.
John Ross
January 5, 2026 AT 05:28Quality system first-no debate. CAPA isn’t a form you fill out after the fact. It’s a living process. If your deviation log looks like a bingo card of recurring issues, you’re not fixing root causes-you’re just playing whack-a-mole with regulators. And don’t even get me started on lab controls. If your stability data isn’t tied to a validated method with traceable instrument logs, you’re not manufacturing drugs-you’re performing interpretive dance for the FDA.
Clint Moser
January 6, 2026 AT 18:58they dont even check the right things. i mean like, why are they focused on training logs when the real issue is the chinese suppliers sending fake API? the fda is so busy checking signatures they miss the whole damn supply chain is rigged. also why do they need 10 copies of every record? its 2026. upload it to the cloud. stop with the paper. i saw a guy get cited for a smudge on a form. smudge. not contamination. smudge.
Ashley Viñas
January 7, 2026 AT 13:02Oh sweetie, if you think mock inspections are enough, you’re already behind. Real readiness means your night shift technician knows why the dissolution limit is 80–120%-not because someone told her to memorize it, but because she understands the pharmacokinetics. And if your quality unit doesn’t have veto power over production? You’re not compliant-you’re just pretending. Also, your SOPs should be written in language a 16-year-old could follow. If it takes a PhD to understand your change control form, you’ve already lost.
Mandy Kowitz
January 7, 2026 AT 18:19So let me get this straight-we’re supposed to be ‘always ready’ for an agency that can’t even count how many opioid pills they’ve lost? The FDA is a joke. They’re just here to make us feel guilty while their own labs can’t even replicate their own tests.
Justin Lowans
January 8, 2026 AT 21:44This is one of the most balanced, practical, and deeply insightful overviews of CGMP readiness I’ve encountered in years. The emphasis on culture over compliance-on ownership over optics-is not just correct, it’s visionary. Many organizations mistake documentation for diligence, but true quality is woven into daily behavior. The PreCheck program, while underutilized, represents a paradigm shift from punishment to partnership. I urge every facility leader to engage with it-not as a checkbox, but as a covenant with patient safety.
Michael Rudge
January 8, 2026 AT 22:08Wow. Just… wow. You wrote an entire novel about paperwork. Can we please talk about the fact that 70% of these inspections happen in India and China, where labor is cheap and corruption is systemic? The FDA doesn’t care if your SOPs are perfect-they care that your CEO didn’t bribe the inspector. But no, let’s all pretend this is about ‘quality culture’ while the real issue is global supply chain exploitation. You’re all just dancing on the edge of a cliff and calling it ‘best practice’.
Doreen Pachificus
January 9, 2026 AT 07:03Interesting. I’ve worked in a facility that had zero 483s for 8 years. We didn’t have fancy software or a big budget. We just had one rule: if you didn’t write it down, it never happened. And if you wrote it down, you had to be able to explain it to a stranger in 30 seconds. The inspectors loved us. Not because we were perfect-but because we were honest. No drama. Just facts.
Akshaya Gandra _ Student - EastCaryMS
January 9, 2026 AT 21:26really helpful but i am student from india and i dont understand all jargon like capa or dmf can someone explain in simple way? also why fda care so much about paper? we dont have so much paper in our lab here. is it same in usa?
en Max
January 11, 2026 AT 20:57Thank you for this comprehensive, meticulously structured overview. The emphasis on systemic integrity-particularly the interdependence of the six quality systems-is both technically accurate and philosophically profound. I would add, however, that the true metric of success is not the absence of 483s, but the presence of psychological safety: when a technician feels empowered to halt production without fear of retaliation, that’s when quality becomes intrinsic. PreCheck is not merely a program-it is an ethical imperative. The FDA’s role, at its core, is not to punish, but to prevent harm. And prevention begins with humility, not compliance.
Angie Rehe
January 12, 2026 AT 01:41Let’s be real-this whole thing is theater. The FDA doesn’t care about your SOPs. They care about headlines. If you’re a small generic maker, they’ll nitpick your training logs to make you look bad so they can push you out of the market and give the big pharma players more shelf space. I’ve seen it. They’ll take your 483, turn it into a warning letter, then quietly approve your competitor’s application two weeks later. It’s not about safety. It’s about consolidation.